Feb 21, 2012
Introduction By William N. Hait, MD, PhD
By William N. Hait, MD, PhD
Johnson & Johnson Pharmaceuticals Research & Development
Soon we will know our susceptibility to all diseases, but will have noidea what to do about it. Genotypic changes plus a life ofenvironmental exposures may (or may not) lead to progression to seriousdisease. In certain instances, we can track progression and interveneappropriately. Oncologic diseases offer an important opportunity forpotentially more effective approaches to therapy, i.e., the treatmentof pre-malignances (“cancer interception,”a phrase coined by Dr. Elizabeth Blackburn of the University ofCalifornia, San Franciso).
Multiple myeloma is a life-threatening disease characterized byend-organ damage, including intensely painful fractures of the bone (afeeling described by one of my patients as “stubbing your toebut it never goes away”). Pre-myelomas are identified byclonal expansion of plasma cells, including monoclonal gammopathy ofundetermined significance (MGUS) and smoldering multiple myeloma (SMM).Dr. Robert Kyle’s excellent review describes the diagnosisand prognosis of these pre-malignant conditions and makes thoughtfulrecommendations regarding management.
Ideally, prevention of disease would entail minimal morbidity and wouldtarget those individuals at greatest risk. MGUS and SMM progress to MMat a predictable rate based on characteristics described by Dr.Kyle’s group that include the quantity of the M-spike anddegree of marrow plasmacytosis.1 Therefore, a group of patients at highrisk for progressing to a fatal disease can be diagnosed andpotentially treated. Might these patients have been spared themorbidity and mortality of multiple myeloma and its treatment byearlier therapeutic intervention?
There are reasons to suspect that early intervention, i.e., thetreatment of pre-malignancies, will be more effective. For example, aBCR-ABL inhibitor such as imatinib is highlyeffective when used to treat chronic myelogenous leukemia (CML) inchronic phase.2 However, as CML progresses to accelerated phase and onto blast crisis, this well-tolerated medication loses almost half ofits efficacy.3
To make cancer interception a reality, much preparatory work needs tobe done. Identification of practical regulatory endpoints, such asvalid surrogates of clinical benefit and metrics for payor acceptance,remain important opportunities for further progress.
Dr. Kyle wisely opines that patients with SMM at low risk forprogression should be carefully observed, and those who are at thehighest risk should not be treated outside of a clinical trial.Preliminary studies with “imids” and dexamethasoneare encouraging.4,5 Recently, anti-interleukin 6 monoclonal antibodies(e.g., siltuximab) have been developed and are undergoing investigationin patients with high-risk SMM, tracked using a newly developedcirculating myeloma cell kit.
Shakespeare wrote in Hamlet, “Disease desperate wrought bydesperate measures are relieved or not at all.” In contrast,the predecessors to disease should require less desperation and greaterconcentration by the biomedical research community. Carefulcharacterization of premalignant conditions exemplified by the work ofDr. Kyle’s group is essential if we are to make cancerinterception a reality.
Dr.Hait, a medical oncologist, is Senior Vice President and WorldwideTherapeutic Area Head of Oncology at Johnson & JohnsonPharmaceuticals Research & Development, LLC. He currentlyserves on ASCO’s Cancer Research Committee.
References
1. Kyle RA, Remstein ED, Therneau TM, et al. N Engl J Med.2007;356:2582-90.
2. Druker BJ, Talpaz M, Resta DJ, et al. New Engl J Med.2001;344:1031-7.
3. Druker BJ, Sawyers CL, Kantarjian H, et al. N Engl J Med.2001;344:1038-42.
4. Detweiler-Short K, Hayman S, Gertz MA, et al. Am J Hematol.2010;85:737-40.
5. Mateos M-V, López-Corral L, Hernández M, etal. Smoldering Multiple Myeloma (SMM) At High-Risk of Progression toSymptomatic Disease: A Phase III, Randomized, Multicenter Trial BasedOn Lenalidomide-Dexamethasone (Len-Dex) As Induction Therapy Followedby Maintenance Therapy with Len Alone Vs No Treatment. ASH AnnualMeeting Abstracts. 2011:Abst 991.
Insights into Smoldering (Asymptomatic) Multiple Myeloma (SMM) By Robert A. Kyle, MD
By Robert A. Kyle, MD
MayoClinic
Smoldering (asymptomatic) multiple myeloma (SMM) is defined by thepresence of a monoclonal (M) protein level ≥ 3 g/dL and/or≥ 10% monoclonal plasma cells in the bone marrow, but noevidence of end-organ damage.1 End-organ damage(“CRAB”) consists of hypercalcemia (C), renalinsufficiency (R), anemia (A), or bone lesions (B) due to the plasmacell proliferative disorder. SMM must be differentiated from monoclonalgammopathy of undetermined significance (MGUS) because of its greaterrisk of progression to multiple myeloma (MM) or a related disorder. Therisk of progression to symptomatic MM is approximately 10% per year forSMM compared to 1% per year for MGUS.
MGUSand disease progression
MGUS is found in 3% of the white population age 50 or older. It is morecommon in men (4.0%) than in women (2.7%). The prevalence increases to5% in persons age 70 or older and to 7.5% among those age 85 or older.The size of the M protein is modest, with more than 60% having an Mprotein < 1.0 g/dL. Uninvolved immunoglobulins are reduced inless than one-third of patients.2 The prevalence of MGUS in blackpatients is approximately twice that of the white population, and theprevalence in Japanese patients is approximately two-thirds that of thewhite population.3,4 This was confirmed in patients from Ghana, wherethe prevalence was 5.8% in 917 men age 50 years or older.5 In contrast,the prevalence of MGUS in Nagasaki, Japan, was 2.4% in patients age 50older.6 It should be pointed out that MGUS precedes virtually all casesof MM.7
It is impossible to know whether a patient with MGUS will remain stableor progress to a plasma cell malignancy such as MM,Waldenström macroglobulinemia, or AL amyloidosis. Predictorsof progression include the size of the serum M protein at the time ofrecognition of MGUS. The risk of progression 10 years after therecognition of MGUS was 6% for those with an M-protein level of 0.5g/dL or less in contrast to 24% for those with an M protein of 2.5g/dL. At 20 years, the risk of progression in a patient with an Mprotein of 1.5 g/dL was 1.9 times the risk of progression with aninitial value of ≤ 0.5 g/dL, while the risk of progression withan M protein of 2 g/dL initially was 4.6 times the risk of progressionwith an initial value of 0.5 g/dL. Patients with an IgM or an IgAmonoclonal protein have an increased risk of progression compared tothose with an IgG protein. The number of bone marrow plasma cells inthe bone marrow is also an important factor. The presence of anabnormal serum free light chain ratio (FLC) is found in about one-thirdof patients with MGUS. The risk of progression in these patients washigher than in patients with a normal FLC ratio (hazard ratio = 3.5).This was independent of the level and type of serum M protein.8
Risk factors consisting of an elevated serum M protein ≥ 1.5g/dL, an IgA or an IgM monoclonal protein, and an abnormal FLC ratiohad a risk of progression at 20 years of 58% (high risk), compared withonly 5% when none of these risk factors were present. It must be keptin mind that death from cardiovascular disease, cerebrovascular events,non-plasma cell malignancies or other causes unrelated to theplasma-cell proliferative process are much more common than death froma plasma cell disorder during long-term follow-up.
Patients with MGUS should not be treated but should be tested again infour to six months to exclude the possibility of an evolving MM. Ibelieve that patients with low-risk MGUS may be reevaluated every twoyears, whereas those with high-risk MGUS should be followed annually oruntil they develop an unrelated condition that significantly limitslife expectancy.
DistinguishingSMM from MM requiring therapy
SMM is a more advanced premalignant stage than MGUS. Just as in MGUS,there is no evidence of CRAB (no end-organ damage) related to theplasma-cell proliferative process. However, patients with SMM mayfulfill theusual diagnostic criteria of MM such as a serum M spike ≥ 3g/dL, 10% or more plasma cells in the bone marrow, reduction ofuninvolved immunoglobulins in the serum, and monoclonal light chains inthe urine. Thus, SMM must be distinguished from MM requiring therapy.Approximately 10% to 20% of patients with newly diagnosed MM actuallyhave SMM.
In a group of 276 patients who fulfilled the criteria for SMM, themedian age of 64 (with only 3% younger than 40) and 62% male weresimilar to that in MM.1 The serum M protein at diagnosis ranged from0.5 to 5.4 g/dL with 11% of the patients having an M spike of ≥4 g/dL. IgG was the most common (74%), while 22.5% had IgA, 0.5% had anIgD monoclonal protein and biclonal gammopathies were found in 3%.Kappa was the most common light chain at 67%, with lambda in theremaining 33%. Reduction of uninvolved immunoglobulins occurred in 83%.A monoclonal light chain was found in the urine in 53% but was <0.1 g/24h in 84%. The most common proportion of bone marrow plasmacells was in the 15% to 19% category. Only 10% had fewer than 10%plasma cells in the bone marrow, while 10% had 50% or more bone marrowplasma cells.
During follow-up, 85% of patients with SMM died. During this period,symptomatic MM developed in 57%, while AL amyloidosis was recognized in2%. The median time to progression (TTP) was 4.8 years. The mediansurvival of the patients who developed MM was 3.4 years, which wassimilar to that of MM during the same period.
Deaths from non-myeloma disorders, including cardiovascular andcerebrovascular disease as well as non-plasma cell cancers, were 18% atfive years, 26% at 10 years, 30% at 15 years, and 35% at 20 years. Theoverall survival of SMM was 60% at five years, 34% at 10 years, and 20%at 15 years (median, 6.3 years).
Riskof progression
The risk of progression of SMM to MM was 10% per year for the firstfive years, 3% per year for the next five years, and then 1% to 2% 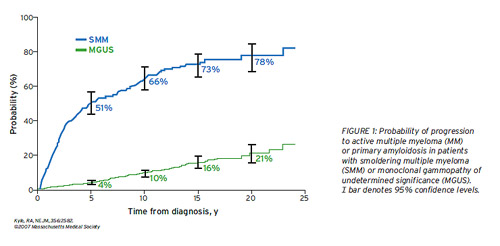 peryear for the next decade (Figure 1).1 This is in contrast to MGUS,which has a risk ofprogression of approximately 1% per year following recognitionthroughout more than 25 years of follow-up.9
peryear for the next decade (Figure 1).1 This is in contrast to MGUS,which has a risk ofprogression of approximately 1% per year following recognitionthroughout more than 25 years of follow-up.9
The risk of progression to active MM or AL amyloidosis at 10 years was55% for patients with an initial plasma cell level of 10% to 14%,compared to progression in 70% of the patients who had more than 50%plasma cell infiltration of the bone marrow. At 10 years, the risk ofprogression to active MM or AL amyloidosis was 57% in patients with aninitial M protein of 2 g/dL and 70% in those with an M protein of 5g/dL. On multivariate analysis, the size of the serum M protein and thenumber of bone marrow plasma cells were the most significantindependent risk factors for progression. The cumulative probability ofprogression at 15 years was 87% in patients with ≥ 10% plasmacells and ≥ 3 g/dL of M protein compared to 70% for those with≥ 10% plasma cells and < 3 g/dL of M protein, and only39% for the patients with < 10% plasma cells and ≥ 3g/dL of M protein. The median TTP was two years in the first group,eight years in the second group, and 19 years in those with fewer than10%bone marrow plasma cells and ≥ 3 g/dL of monoclonal protein. Atfive years of follow-up, 66% of patients with IgA experienced diseaseprogression, compared to 46% with IgG. At 10 years, 77% of patientswith IgA experienced disease progression, compared to 62% with IgG. Inaddition to the size of the M protein and number of bone marrow plasmacells, an FLC ratio of ≤ 0.125 or ≥ 8 was anindependent risk factor for progression. Incorporating the FLC ratiointo the risk model, the five-year progression rates in those with bonemarrow plasma cells ≥ 10% and a serum M protein ≥ 3g/dL was 76%, while those with bone marrow plasma cells ≥ 10%but a serum M protein < 3 g/dL was 51%. The risk of progressionwas only 25% for those with a serum M protein ≥ 3 g/dL, bonemarrow plasma cells < 10%, and an FLC ratio of > 0.125 to< 8.10
Treatmentof patients with SMM
I believe that patients with SMM should be observed for evidence ofprogression and not treated unless they are part of a clinical trial.The blood tests should be repeated two to three months after theinitial recognition to exclude the possibility of an evolving MM. Ithas been suggested that there are two types of SMM—anevolving SMM characterized by progressive increase of the serum Mprotein until symptomatic MM develops and a non-evolving SMM in whichthe M protein is stable and then abruptly increases when symptomatic MMdevelops.11 If stable, testing should be repeated every four to sixmonths for the first year and if still stable, reevaluate at six- to12-month intervals.
Efforts to treat patients with SMM have been reported. In a series of29 eligible patients with SMM, 34% had a partial response tothalidomide. The median TTP to symptomatic myeloma was 35 months. Themedian TTP was 61 months for those achieving a partial response, 39months for those with a minimal response (MR), and nine months forthose whose disease failed to respond.12 Mateos et al. reported that118 patients with SMM at high risk of progression were randomlyassigned to lenalidomide and dexamethasone or no treatment. Fourpatients experienced disease progression in thelenalidomide/dexamethasone regimen, compared to 28 of 61 (46%) in theplacebo arm. The three-year overall survival was 98% in the treatedpatients, compared to 82% for placebo.13
In my opinion, the key is to recognize the patients with SMM at thehighest risk for progression and then treat them in a clinical trialwith a regimen active for multiple myeloma and with as few side effectsas possible. One would like to identify those patients who are at a 90%risk of progression at two years and treat them in a randomizedclinical trial.
Dr.Kyle is a Professor of Medicine, Laboratory Medicine, and Pathology atMayo Clinic and has been an ASCO member since 1968. An internationallyrecognized expert in hematologic malignancies, he was the first todescribe monoclonal gammopathy of undetermined significance andsmoldering multiple myeloma. In 2007, Dr. Kyle was presented with theDavid A. Karnofsky Memorial Award, ASCO’s highest scientifichonor.
References
1. Kyle RA, Remstein ED, Therneau TM, et al. N Engl J Med.2007;356:2582-90.
2. Kyle RA, Therneau TM, Rajkumar SV, et al. N Engl J Med.2006;354:1362-9.
3. Cohen HJ, Crawford J, Rao MK, et al. Am J Med. 1998;104:439-44.[Erratum. Am J Med. 1998;105:362.]
4. Landgren O, Gridley G, Turesson I, et al. Blood. 2006;107:904-6.
5. Landgren O, Katzmann JA, Hsing AW, et al. Mayo Clin Proc.2007;82:1468-73.
6. Iwanaga M, Tagawa M, Tsukasaki K, et al. Mayo Clin Proc.2007;82:1474-9.
7. Landgren O, Kyle RA, Pfeiffer RM, et al. Blood. 2009;113:5412-7.
8. Rajkumar SV, Kyle RA, Therneau TM, et al. Blood. 2005;106:812-7.
9. Kyle RA, Therneau TM, Rajkumar SV, et al. Mayo Clinic Proc.2004;79:859-66.
10. Dispenzieri A, Kyle RA, Katzmann JA, et al. Blood. 2008;111:785-9.
11. Rosiñol L, Bladé J, Esteve J, et al. Br JHaematol. 2003;123:631-6.
12. Detweiler-Short K, Hayman S, Gertz MA, et al. Am J Hematol.2010;85:737-40.
13. Mateos M-V, López-Corral L, Hernández M, e tal. Smoldering Multiple Myeloma (SMM) At High-Risk of Progression to Symptomatic Disease: A Phase III, Randomized, Multicenter Trial Based On Lenalidomide-Dexamethasone (Len-Dex) As Induction Therapy Followed by Maintenance Therapy with Len Alone Vs No Treatment. ASH Annual Meeting Abstracts. 2011;Abst 991.