
As we are all aware, lately there has been a call for “personalized, precision medicine.” Assuming you are a caregiver, I suspect you have been trying to provide personalized medicine as precisely as you can for your entire career. This concept is not new, arguably dating to Hippocrates and beyond. Indeed, Virchow introduced the field of microscopic pathology in the 1870s,1 Roentgen began the field of radiographic imaging in the 1890s,2 Bloom and Richardson proposed tumor grading in the 1950s,3 and the introduction of CT, MRI, and PET scans in the 1970s, ‘80s, and ‘90s represented major steps forward in the care of our patients. Using breast cancer as a more specific example, in the 1970s, McGuire and others demonstrated that estrogen receptor (ER) status was highly predictive of benefit from anti-estrogen therapy; more recently, tests for HER2 amplification or overexpression appear to be pretty good “companion diagnostics” for therapeutic agents directed towards this target (Fig. 1).4,5
So what’s the problem? Frankly, none of these diagnostics had to go through prospective randomized trials to demonstrate their relative worth in clinical care—rather, the benefit of applying these diagnostics simply appeared to be intuitive. However, it is no longer that easy. The advent of rapid and relatively affordable high-throughput molecular analyses (genomics, transcriptomics, proteomics, metabolomics, etc.) has permitted development of a bewildering next generation of new cancer diagnostic tests that are being introduced into clinical care using our grandparents’ regulatory structure.
What is that structure? Most clinicians, thankfully, have not had to be aware of the inconsistent regulatory environment in the United States for introduction of diagnostic tests into their practice. We could spend our time in better ways, like taking care of our patients. This is no longer the case. If we are going to use tumor biomarker tests to drive our therapy, and we should, then we need to value tumor biomarker tests in the same way we value drugs.6 Therefore, it’s time for a brief primer.
Spoiler alert: this gets wonky. The U.S. Food and Drug Administration (FDA) regulation of medical products is basically divided into two arenas: one to evaluate new drugs and one to evaluate new devices. As most of us are aware, new therapeutics must be proven to be “safe and effective” before they can be offered to patients. The definitions of safe and effective vary, but in general no drug can be marketed without first receiving FDA approval. The review of a new drug occurs, not surprisingly, in the Center for Drug Evaluation and Research (CDER) and cancer drugs are the responsibility of the Office of Hematology and Oncology Products (OHOP). For oncology drugs, although not absolutely required, the FDA has a standing external Oncologic Drugs Advisory Committee (ODAC) consisting of physicians, statisticians, lay advocates, and others, that is available to provide review and guidance as OHOP considers a new drug. The decision to approve a new drug is based on high levels of evidence that it is, in fact, safe and effective, and third-party payers mostly follow FDA recommendations when they determine coverage. This system is not perfect and results in occasional controversy, but it has generally led to an armamentarium of highly effective anticancer agents with predictable toxicities, accompanied by a remarkable reduction in cancer mortality and morbidity over the last few decades.
On the other hand, cancer diagnostics are not regulated in such a straightforward manner, if they are regulated at all. In fact, there are two avenues for introducing diagnostic tests into clinical use, including those of tumor biomarkers (Fig. 2). Dating back to the 1970s, with the explosion of antibody-based diagnostic tests, the FDA made an enforcement discretion decision to permit development and use of diagnostic tests in individual laboratories without any requirement that they be reviewed by the agency. These assays were designated as laboratory-developed tests (LDTs), defined by the FDA as an in vitro diagnostic that is intended for clinical use and designed, manufactured, and used within a single laboratory. Within this enforcement discretion, an LDT could be provided to clinicians for patient decision making, as long as they were performed in laboratories approved under the Clinical Laboratory Improvement Amendments (CLIA), which are administered not by the FDA but by the Centers for Medicare and Medicaid Services (CMS). Results from an LDT can be shared with a provider, and submitted for reimbursement, as long as the tests are not sold to second parties to perform the assays.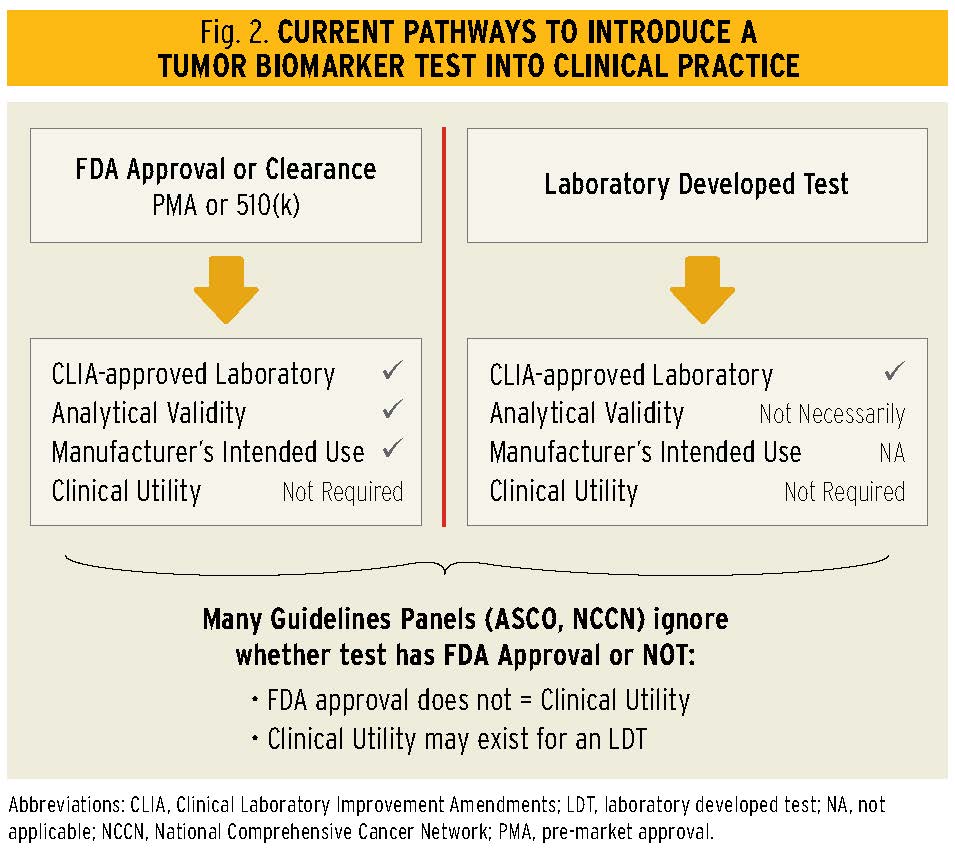
Alternatively, if one wishes to market a test or test kit, FDA review is required. If the test is brand new, it is submitted for pre-market approval (PMA), whereas if it is a new kit but based on a previously existing, approved test, it is considered for clearance following submission of a 510K application. Tumor biomarker tests are evaluated by the Office of In Vitro Diagnostics and Radiological Health (OIR), which is within the Center for Devices and Radiological Health (CDRH).
Now, for those of us who are clinicians, here’s where the rubber meets the road. In general, most oncologists depend on the FDA, along with groups that provide evidence-based pathways and guidelines (such as ASCO and the National Comprehensive Cancer Network), to determine when and if a drug should be offered to their patients. Sure, there is wiggle room to vary a bit from these guidelines, but at least a clinician can trust that the drug has been very carefully evaluated by experts in the field, and it is reasonably likely to be safe and effective and to offer some measure of clinical benefit to a patient.
Unfortunately, such is not the case for tumor biomarker tests. Nearly a decade ago, Teutsch et al., writing for the Evaluation of Genomic Applications in Practice and Prevention (EGAPP) proposed three semantic terms that more carefully articulated some of the problems that evolved from this regulatory duality:
1) Analytical validity. This basically means that the test is a well-performed and highly accurate test, taking into account pre-analytical issues (e.g., was the specimen processed in time and appropriately for the test in mind?) and analytical issues (e.g., is it accurate, sensitive, specific, and reproducible?)
2) Clinical validity. Does the assay divide one population into at least two or more that differ either biologically or clinically with statistical significance?
3) Clinical utility. Defining the latter term is the key issue, and there is not consensus on a clear definition or on the level of evidence required to demonstrate clinical utility. However, to me, clinical utility implies that there are high levels of evidence that use of the assay for the biomarker supports a treatment decision that produces a better outcome for the patient than if the marker results were not available—preferably better overall survival or quality of life, or both—or equal outcomes with lower cost or inconvenience (which, theoretically, should be factored into the quality-of-life assessment anyway).
Remember, a tumor biomarker is not a test—rather, it is a marker of some perturbation from normal that indicates malignant behavior. There may be one or more tests (or assays) for a tumor biomarker, done in one or more ways, leading to enormous heterogeneity in the results that we, as clinicians, may receive. Back to our drug analogy. Would you use a drug if you weren’t sure of how it was prepared, mixed, or infused? I suspect not. Would you recommend a treatment if there were no data to suggest it improves the patient’s outcomes? Again, I hope not. Yet, because of the inconsistent regulatory environment, the tests that you use to make critical decisions to better personalize the care of your patients with precision may be performed using a commercially available, FDA-approved kit exactly as the manufacturer recommends, or using the same kit but “tweaked” in unknown ways by the laboratory doing the test, or using reagents and methods that are presumably accurate but that have never been reviewed for analytical validity (in other words, using an LDT) by the FDA.
Further, even FDA approval or clearance does not mean the test has clinical utility. Although the FDA does not use this terminology, the OIR, by legislative fiat, reviews analytical validity very carefully, but may approve a new or predicate test if it merely meets the manufacturer’s claim for an intended use. While this intended use may equate to clinical validity, it may or may not result in better outcomes for the patient if applied in the clinic.
So what does all this mean? Great tests may or may not ever be reviewed by the FDA, and tests with no clinical utility may be marketed as FDA approved or cleared. Indeed, since its inception, the ASCO Tumor Marker Guidelines Expert Panels have taken the stance that their recommendations for use of a given tumor marker test will be based on whether there are high levels of published evidence of analytical validity and clinical utility, regardless of whether the test has been approved, cleared, or never reviewed by the FDA.7
All of this wonkiness raises a final question: how are high levels of evidence for a tumor biomarker test generated? A few years ago, I had the privilege of co-authoring a set of proposals with two distinguished colleagues: Dr. Richard Simon of the National Cancer Institute, long considered one of the fathers of cancer biostatistics, and Dr. Soon Paik, who has led several groundbreaking and practice-changing pathology correlative studies within the National Surgical Adjuvant Breast and Bowel Project (NSABP).8 We proposed that high levels of evidence for a tumor biomarker test could be generated in one of two ways. One of these is the classical approach, as is required for drug approval, by performing a prospective randomized clinical trial (PRCT), in which clinical utility of a biomarker test is the primary objective of the trial itself. Various designs for such trials have been suggested by Sargent et al.9 and Freidlin et al.10 However, precious few PRCTs addressing the biomarker itself (such as the TailoRx, RxPonder, MINDACT, and SWOG S0500 trials) have been conducted, because they are time-consuming and costly.
Unlike drugs, an alternative path to a PRCT is available for generation of high levels of evidence for a tumor biomarker test: a “prospective retrospective” study.8 In such a study, specimens collected and archived within prospectively conducted clinical trials that addressed a clinically important question can be studied for a factor that might have clinical utility for that question. For example, one might use archived specimens from adjuvant trials in which patients with HER2-positive breast cancer were randomly assigned to trastuzumab or not to determine if a new tumor biomarker test, for which analytical and clinical validity are already available so that the test is locked down, predicts who did or did not benefit from the therapy.
What’s the answer to all this chaos? ASCO is very supportive of efforts within the FDA to reform tumor biomarker review and use. Recently, the FDA has announced that the Agency will establish Oncology Centers of Excellence. Although it is not clear what will arise from this reorganization, one hopes it will lead to consistent evaluation of both drugs and devices related to cancer care, with something like the existing ODAC to provide some level of corporate memory and external expertise from one situation to the next for tumor biomarker tests as it presently does for drugs.
Second, the FDA has issued a Guidance Document proposing a very thoughtful and measured phase out of the enforcement discretion stance that permits LDTs to be introduced into clinical use without FDA oversight. FDA has not proposed elimination of all LDTs in all of medicine or even that all LDTs will require FDA review. Rather, the Agency will take a “risk-based approach to regulatory oversight of LDTs [that] is appropriate and necessary to protect patient safety.” while continuing to use enforcement discretion for “LDTs used solely for forensic (law enforcement) purposes, certain LDTs for transplantation…Low-risk LDTs (Class I devices), LDTs for rare diseases and ‘Traditional LDTs’, and ‘LDTs for Unmet Needs, when no FDA-approved or cleared equivalent device is available.”
This latter approach has been controversial.11 Institutional, academic, and private laboratories have argued that such a move will stifle innovation and reduce availability of laboratory tests to patients by placing regulatory and bureaucratic hurdles in their way.
On the other hand, the current inconsistent regulatory environment creates uncertainty and, indeed, a level of chaos in what has become, more or less, based on clinical caveat emptor. Further, commercial firms that develop highly analytically validated tumor biomarker assays—and, more importantly, support the rigorous studies necessary to demonstrate clinical utility—find their rewards for doing so diluted by CLIA-approved laboratories developing LDTs that are purportedly, although not proven, equivalent to the commercial test.
ASCO is very supportive of the FDA Draft Guidance Document to revise the enforcement discretion decision and require FDA review of high-risk tumor biomarker tests. We feel that although the LDT approach has been of value over the last 4 decades, it is time to recognize the absolute importance that tumor biomarker tests now have in modern oncology in directing cancer treatment, whether they be individual analytes such as ER or HER2 (or many others now proven to be important), multiparameter signature tests, panel assays that include several individual analytes in the same overall suite of tests, or, in some cases, true next-generation sequencing.
It is time we place the same value on these critical diagnostics as we do on drugs, so that we have confidence in both the analytical validity and the clinical utility of tests that we will use to treat, or perhaps more importantly, to withhold treatment with lifesaving but toxic and expensive antineoplastic agents. Our patients deserve nothing less.
References
1. Virchow R. The Croonian Lecture on the Position of Pathology among the Biological Studies: Delivered before the Royal Society, March 16th, 1893. Br Med J. 1893;1:561-5.
2. Roentgen WC. [On a new kind of ray (first report)]. Munch Med Wochenschr. 1959;101:1237-9.
3. Bloom HJ, Richardson WW. Histological grading and prognosis in breast cancer; a study of 1409 cases of which 359 have been followed for 15 years. Br J Cancer. 1957;11:359-77.
4. Hammond ME, Hayes DF, Dowsett M, et al. American Society of Clinical Oncology/College Of American Pathologists guideline recommendations for immunohistochemical testing of estrogen and progesterone receptors in breast cancer. J Clin Oncol. 2010;28:2784-95.
5. Wolff AC, Hammond ME, Hicks DG, et al. Recommendations for human epidermal growth factor receptor 2 testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists clinical practice guideline update. J Clin Oncol. 2013;31:3997-4013.
6. Hayes DF, Allen J, Compton C, et al. Breaking a vicious cycle. Sci Transl Med. 2013;5:196cm6.
7. Hayes DF, Bast RC, Desch CE, et al. Tumor marker utility grading system: a framework to evaluate clinical utility of tumor markers. J Natl Cancer Inst. 1996;88:1456-66.
8. Simon RM, Paik S, Hayes DF. Use of archived specimens in evaluation of prognostic and predictive biomarkers. J Natl Cancer Inst. 2009;101:1446-52.
9. Sargent DJ, Conley BA, Allegra C, et al. Clinical trial designs for predictive marker validation in cancer treatment trials. J Clin Oncol. 2005;23:2020-7.
10. Freidlin B, McShane LM, Korn EL. Randomized clinical trials with biomarkers: design issues. J Natl Cancer Inst. 2010;102:152-60.
11. Evans BJ, Burke W, Jarvik GP. The FDA and genomic tests--getting regulation right. N Engl J Med. 2015;372:2258-64.
Comments
Dane J. Dickson, MD
Dec, 26 2016 12:26 PM
Dan's article is masterful and crucial. Until we tie biomarkers and clinical outcomes directly together, we will never be able to trust the power of these tools. Standardization and comparison of LDTs and tying these back to treatments and resulting outcomes is the responsibility of many groups. Randomized clinical trials with biomarkers are generally not feasible for cost reasons, and furthermore the science is advancing so quickly that many of these biomarkers are good right now, but should be iteratively improved. In otherwords, biomarkers as dynamic tools that must improve over time - (think of HER-2 testing - what would have happened if we waited for perfection before he fully introduced it to the marker), A very fasible alternative is the Prospective Obersvational Outcome Registry tied to testing standardization. As information is learned, the testing can be improved in an iterative approach.
Daniel F. Hayes, MD, FASCO, FACP
Dec, 28 2016 2:21 PM
Thanks for thoughtful comments!
L. Michael Glode, MD, FACP, FASCO
Dec, 30 2016 5:13 PM
Dan, your article is a superb overview. Having lived with PSA for 30 years, the prostate cancer field could provide an excellent test for all sorts of retrospective analyses of policy decisions, costs associated with them, and the pitfalls of early application. When first introduced, many urologists were heard to consider it a "Providentially Sent Antigen" in meetings I attended. Its widespread use in screening went unchecked with the unchallenged (for the most part) belief that finding cancer earlier would result in saving lives. Only decades later did the truth emerge that the "lives saved" would need to be weighed against the morbidity of potentially unnecessary treatment for a more balanced view of PSA screening. In the prostate cancer field, we now find ourselves facing a repeat of this situation with nearly a dozen biomarkers that may improve analysis of risk, but are far from satisfying the more stringent criterion that acting upon such results will result in improved patient outcomes. Rather, the rush to commercialization more typically evaluates whether physicians "change their mind" or "recommend an alternative therapy" when presented with a given test result. Striking a balance in the approval of a new test versus the understandable desire for market success is woefully needed. Iteration may be the answer, although we would need to accept the disapproval of some tests (or their inappropriate use) as part of the bargain. Your article is a great start - congratulations!
Daniel F. Hayes, MD, FASCO, FACP
Dec, 31 2016 1:42 PM
Thanks mike. Sadly, FDA withdrew their draft guidance to clarify the LDT decision last month so we're back to current chaos. And I agree- giving doctors information and the seeing if they act on it is a test of human nature, not worth of the information itself!
David Barnes
Jan, 22 2017 6:58 PM
The expensive and time consuming approval process for drugs is justified based on unintended and potentially fatal side effects. This stems directly from the fact that drugs interfere with biological processes. Herein lies the fundamental distinction between drugs and tumor markers. Tumor markers interrogate biological processes without interfering with them. Of course, it is important to recognize that the results produced by tumor markers can be used to guide the use of drugs. But for there to be a strong case to be made for an approval path for tumor markers that was like the path for drugs, a much stronger case would have to be made for the shortcomings of the existing process and potential for harm. Otherwise, an approval frame work that was prohibitively expensive and time consuming would be created without justification.
The FDA drug approval process has resulted in a landscape of oncology drugs characterized by marginal efficacy and substantial toxicity. Tumor markers offer hope of improving this landscape for the benefit of patients. Maintaining an expedient risk based approval process for tumor markers is essential to continued momentum.