Jul 17, 2018

This issue’s “Current Insights in Oncology” is excerpted from the 2018 ASCO Educational Book, an NLM-indexed collection of articles written by ASCO Annual Meeting faculty and invited leaders from ASCO’s meetings. Published annually, each volume of the ASCO Educational Book highlights the most compelling research and developments across the multidisciplinary fields of oncology.
By John D. Hainsworth, MD, and F. Anthony Greco, MD
Cancer of unknown primary site (CUP) is a clinical syndrome that includes many different cancer types and accounts for approximately 2% of all cancer diagnoses. Although the anatomic primary sites in patients with CUP cannot be identified clinically, they are identified in approximately 75% of postmortem examinations, and most are less than 1 cm in size.1,2 The biologic mechanisms underlying this unique clinical behavior (i.e., dissemination of cancer while the primary site remains small) is unknown; to date, no specific molecular signatures have been associated with these cancers.
Patients should be diagnosed with CUP only after specific clinical and pathologic studies have been completed.3 Clinical evaluation includes complete history and physical examination, complete blood counts, serum chemistries, urinalysis, CT scans of the chest/abdomen/pelvis, mammogram (women), and serum prostate-specific antigen (men). Pathologic evaluation includes histologic examination and selected immunohistochemical (IHC) stains. When these evaluations are used to define CUP, detection of an anatomic primary site at any time during the subsequent clinical course is uncommon (< 10%).
Treatment of patients with CUP initially is dependent on identification of favorable subsets of patients with specific clinical and/or pathologic presentations.3 These patients (15% to 20% of all patients with CUP) respond relatively well to specific therapies, and some have potentially curable cancers. The remaining 80% to 85% of patients with CUP have traditionally received empiric chemotherapy with regimens designed to have some efficacy in a broad spectrum of cancer types (e.g., taxane/platinum, gemcitabine/platinum).3-6
As treatment improves and becomes more type-specific for many advanced cancers, the notion that empiric chemotherapy can provide adequate therapy to a heterogeneous population of patients with many different cancer types becomes increasingly outdated. Non–small cell lung cancer (NSCLC) and colorectal cancer, both common postmortem diagnoses in CUP series, illustrate the problems involved in empiric treatment. Fifteen years ago, treatment of advanced NSCLC with paclitaxel/platinum (a commonly used empiric CUP regimen) would have provided reasonable treatment. Today, 13 additional drugs are approved for treatment of NSCLC, none of which is approved (or routinely used) in the empiric treatment of CUP. In the treatment of advanced colorectal cancer, even first-line empiric CUP regimens, such as taxane/platinum, are not optimal, and none of the 10 other drugs approved for this indication is used.
The era of precision medicine in oncology offers promise for improved diagnosis and better therapy for patients with the CUP syndrome, and first steps have already been taken toward incorporating precision medicine into the routine management of disease in these patients. This brief review examines new diagnostic methods available for detection of the specific cancer type in these patients. Next, precision treatment for patients with CUP, guided by molecular identification of the cancer type and the detection of actionable molecular alterations, is discussed.
Diagnosis of CUP in the Era of Precision Medicine
The initial evaluation of CUP has always been predicated on the assumption that identification of a primary site or specific cancer type can improve the efficacy of treatment. The questions underlying this assumption have been difficult to address: (1) Do patients with unknown primary site actually have a primary site? The fact that most patients have small primary sites found at autopsy provides strong affirmative evidence. (2) Do CUPs mirror their counterparts that present with overt primary sites in most aspects of tumor biology, even though they differ in the ability to metastasize widely (mechanism unknown) while the primary tumor remains small? (3) Do CUPs respond to the same treatments proven effective in patients with the corresponding cancers of known primary site? Until recently, testing of the second and third questions was indirect, because most patients with CUP never have anatomic primary sites identified. During the past 10 years, improved diagnosis has provided much more information with which to address these questions.
IHC Staining
IHC staining has been part of the standard pathologic evaluation in CUP for the past 20 years. During that time, stains of increased specificity have been developed. Although standard practice varies, most pathologists use panels of IHC stains to narrow the diagnostic spectrum; use of additional stains is guided by the histology, clinical presentation, and results of the initial IHC panel.7
With current IHC staining approaches, a single diagnosis is predicted in 30% to 40% of patients with CUP.8-10 However, there has been reluctance to use IHC diagnoses as a guide for treatment, because the pathology report usually is somewhat equivocal and uses words and phrases like “favor” or “consistent with” rather than giving a firm diagnosis. Until recently, there has been no other method with which to test the reliability of the IHC results.
Gene Expression Profiling
Specific gene expression profiles are now recognized in most cancers according to their site of origin, which reflects the different expression profiles present in their normal tissues of origin.11 The application of these findings to cancer diagnosis was first demonstrated when differences in gene expression allowed the distinction of acute myeloid leukemia from acute lymphoblastic leukemia.12 Differences in gene expression also allow distinction between various solid tumors and provide a valuable method for diagnosis of the tissue of origin in patients with CUP. It is important to recognize that this molecular analysis, which detects patterns of gene expression unique to the tissue of origin, is different from molecular mutation profiling (discussed in the next section), which is designed to detect oncogenes and other actionable molecular alterations but which only rarely determines the cancer type.
Gene expression profiling assays with either reverse-transcriptase polymerase chain reaction or gene microarray techniques are now commercially available. These assays, termed molecular cancer classifier assays (MCCAs), are able to identify more than 40 different cancers and cancer subtypes. In validation studies in cancers of known primary sites (biopsies from primary and metastatic sites), these assays correctly identified the tumor type in more than 85% of cases.11,12 In a group of 252 patients with CUP whose diseases were studied prospectively, primary sites were predicted in 247 (98%) by using the 92-gene CancerTYPE ID assay; only five patients had unclassifiable gene expression profiles.13 As with IHC, the accuracy of these diagnoses is difficult to establish, because anatomic primary sites usually are not identified. However, in a group of 24 patients with CUP who had a primary site identified 2 to 79 months after an initial diagnosis, the correct primary site was predicted by MCCA in 18 (75%) of 24 patients who had adequate tissue available for analysis.14,15
The accuracy of IHC staining compared with MCCA diagnoses has been studied in several trials. In two trials, pathologists were blinded to the tumor type and performed IHC stains (as many as they thought necessary) compared with MCCA in patients with metastatic carcinoma of known primary.9,16 In both studies, the MCCA was more accurate; differences were accentuated when tumors were poorly differentiated (83% vs. 67% accurate in poorly differentiated carcinomas).16 In patients with CUP, the predictive accuracy of IHC (i.e., prediction of a single site of origin) decreases to 30% to 40%.8-10 In the largest study of 149 patients with CUP, IHC results performed at the time of initial evaluation were compared with MCCA results obtained later with remaining tumor tissue.8 IHC evaluation resulted in the prediction of a single site of origin in 35%. In these patients, MCCA results matched the IHC results in 77% of patients. MCCA predicted the primary site in most of the remaining 65% of patients when IHC gave nonspecific results.
Gene expression profiling also results in a high percentage of diagnoses in the uncommon group of patients with poorly differentiated neoplasm of unknown origin. In a group of 30 such patients without a lineage diagnosis after complete pathologic evaluation (median, 18 IHC stains performed), MCCA established the tumor lineage in 25 (83%) of 30 patients and a specific diagnosis in the 10 patients with carcinomas.17 Results with MCCA provide strong evidence that most CUPs retain gene expression profiles similar to their tissue of origin and suggest that responses to site-specific treatment may also mirror their counterparts with known primary site.
Treatment of CUP in the Era of Precision Medicine
For the majority of patients with CUP, empiric combination chemotherapy traditionally has been considered the standard first-line therapy. Benefits of this approach are modest: standard regimens produce response rates less than 40%, median survival of fewer than 11 months, and 2-year survival less than 20%.4-6 Because the site of origin can be determined now in most patients with CUP, site-specific treatments that are based on these molecular diagnoses have been investigated for several years, and these data are reviewed.
For many advanced cancer types, standard treatment now includes the identification of patient subsets defined by the presence of actionable molecular alterations (e.g., HER2 in breast cancer; EGFR/ALK/ROS1 in NSCLC). Analogous molecular testing and targeted treatment of patients with CUP whose cancer types have been diagnosed should also be considered; data to support this approach are developing. Beyond testing for specific molecular alterations that are based on the cancer type, increasing evidence indicates that comprehensive genetic profiling of tumors can identify additional targetable molecular alterations in substantial numbers of patients with advanced cancer. Although evidence in patients with CUP is still limited, these issues are also discussed.
Site-Specific Therapy by Molecular Cancer Classifier Assay Diagnosis
Evidence supporting the use of site-specific therapy directed by the MCCA results is incomplete but is increasingly compelling. Although results of a randomized comparison of site-specific therapy and empiric chemotherapy has not been reported, the available data strongly suggest that outcomes are improved with site-specific therapy, particularly for the more responsive cancers.18-20
The largest prospective trial to date included 194 previously untreated patients with CUP who received site-specific therapy on the basis of a MCCA diagnosis.18 The median survival of all patients was 12.5 months; patients predicted to have responsive tumor types had significantly longer median survival than those with less responsive types (13.4 vs. 7.6 months). Although patient numbers in specific tumor groups were relatively small, survival generally mirrored the expected survival of patients with the predicted cancer types (biliary, 7 months; pancreas, 8 months; colon, 13 months; ovarian, 30 months; and breast, not reached at > 24 months).
Other recent studies also support the use of site-specific treatment on the basis of MCCA results. In one study, a MCCA resulted in a diagnosis in 188 (87%) of 216 patients with CUP.19 Treatment received by 114 patients was examined retrospectively: patients who received treatment predicted effective for their MCCA diagnosis had a median survival of 13.6 months compared with a 6-month median survival for those who received empiric treatments predicted ineffective. Another trial used an empiric regimen of carboplatin/paclitaxel/everolimus; the 18 patients predicted by MCCA to have tumor types sensitive to this regimen had a median survival of 17.8 months, but the 19 patients with tumor types predicted to be insensitive had a median survival of 8.3 months.20
Additional retrospective studies focusing on specific tumor types also support this approach. In three studies, patients with CUP who were predicted to have metastatic colorectal cancer (all had negative colonoscopies) had median survivals of more than 20 months when treated with colorectal cancer therapies.21-23 In a group of 20 patients predicted to have renal cell carcinoma (none had renal lesions on CT scan), site-specific treatment with targeted agents resulted in a median survival of 16 months.3,24 Patients predicted to have germ cell tumor, lymphoma, and neuroendocrine tumors have had typical responses to site-specific therapy for these tumor types.3,17
When patients with CUP are treated with site-specific treatment, it is reasonable to test a tumor biopsy specimen for specific molecular alterations on the basis of the predicted site of origin by using either specific hotspot genetic testing or comprehensive molecular profiling. Examples include testing for HER2 after the diagnosis of breast/gastroesophageal junction/gastric cancer; testing for EGFR/ALK/ROS1 after the diagnosis of NSCLC, and testing for KRAS/microsatellite instability (MSI) after the diagnosis of colorectal cancer. At present, only anecdotal reports document responses to targeted treatment in these patients (e.g., NSCLC with EGFR mutation responding to gefitinib; NSCLC with ALK rearrangement or MET amplification responding to crizotinib; HER2-positive breast cancer responding to trastuzumab).18,25-30 Additional studies in this area are needed.
The rapid development of checkpoint inhibitors and other immunomodulatory agents creates additional possibilities for treatment in patients with CUP. At present, there are only a few case reports about these treatments.31,32 Studies in patients with CUP who are predicted to have potentially sensitive tumor types (e.g., lung, urothelial, renal) are indicated.
Comprehensive Molecular Profiling in CUP
The role of comprehensive molecular profiling is evolving rapidly. Already, the increased use of comprehensive profiling has enabled the testing of targeted agents in a wide variety of advanced cancer types that are rare or have a low incidence of the critical mutations. Not surprisingly, effective targeted agents have activity across a spectrum of tumor types, as long as the critical molecular alteration is present.33-36 For example, HER2 amplification/overexpression predicts response to HER2-targeted therapy in colorectal carcinomas, salivary gland carcinomas, and others, in addition to the cancer types for which HER2-targeted therapy is currently labeled.33 Presence of the BRAF V600E mutation predicts response to BRAF-targeted drugs in NSCLC, ovarian cancer, and others.33,35
The efficacy of targeted agents varies widely on the basis of solid tumor type. The most dramatic example of this is the high response rate of metastatic BRAF V600E–mutated melanoma (>60%) when treated with BRAF inhibitors,37 and the inactivity of the same agents in BRAF V600E–mutated colorectal cancer.38 Therefore, it is unlikely that most of the current targeted agents will ever be recommended for use agnostic of tumor type. However, the U.S. Food and Drug Administration recently approved the first treatment on the basis of molecular testing alone: pembrolizumab for patients with MSI unstable tumors regardless of the cancer type.39 A new targeted drug, larotrectinib (a pan-TRK inhibitor), is likely to become the second such drug when it gains approval for patients with the uncommon TRK mutation.40
Comprehensive molecular profiling of patients with CUP indicates that a substantial number of potentially important molecular alterations are present. In a group of 200 patients with CUPs (125 with adenocarcinoma, 75 with carcinoma), potentially actionable mutations were identified in 169 (85%).34 Some of these tumors had mutations for which only investigational drugs (with undefined activity) are available, and some had mutations that may affect treatment decisions but for which no specific treatment is available (e.g., KRAS). However, 38 (18%) of 200 tumors had molecular alterations for which approved targeted agents are currently available (HER2, BRAF, EGFR, ALK, RET, BRCA, and ROS1).
Recently, ctDNA has been evaluated in 442 patients with CUP; previously characterized molecular alterations were identified in 66% of tumor specimens.31 The actionable mutations identified were similar to those previously reported from tests of CUP tumor tissue.34 Additional validation of the role of ctDNA testing is required, but such testing may have advantages compared with tissue testing.
As biomarkers predictive of response to immune checkpoint inhibitors are identified, it appears that the use of these agents in CUP holds promise, particularly because many of the cancer types identified in the CUP population are responsive to these drugs. IHC staining for PD-L1 has been of some value, but it is not strongly predictive. In a group of 70 patients with CUPs, 63% had IHC staining for PD-1 in tumor-infiltrating lymphocytes, and 21% had cancer cell staining for PD-L1.41 MSI and mismatch repair deficiency are associated with high response rates to checkpoint inhibitors in colorectal cancer and other tumors; pembrolizumab is now approved for advanced cancers with high MSI. The frequency of MSI and mismatch repair deficiency in CUP has not been well studied. High tumor mutation burden also has been associated with higher response rates to checkpoint inhibitors.42 Compared with other tumor types, high tumor mutation burden (≥20 mutations/mb) appears to be frequent in CUP, and the incidence varies with histology: adenocarcinoma, 8%; carcinoma, 11%; and squamous carcinoma, 23%.43
The use of comprehensive molecular profiling to identify and direct therapy for CUP therefore has the potential to contribute substantially to the management of disease in these patients. At present, successful targeted treatment that is based on findings at molecular profiling has been described in anecdotal reports, but no reported prospective clinical trial has evaluated this approach. Therefore, off-study use of targeted treatment presents reimbursement challenges. Until more data exist, results from an MCCA may be helpful; for example, a patient with CUP who has an EGFR mutation and a cancer type identified as NSCLC by MCCA likely will have insurance coverage for therapy with EGFR inhibitors.
A multinational randomized study is needed to address the role of comprehensive molecular profiling in directing treatment of patients with CUP. The ongoing National Cancer Institute MATCH study and the ASCO TAPUR Study will provide some additional information in patients with many advanced cancer types.
Integrated Approach to Diagnosis and Management of CUP
A new approach to the diagnosis and management of the patient with CUP is shown below in Fig. 1. Because diagnosis of the site of origin is now possible in most patients with CUP, initial evaluation closely parallels the strategy used in the initial evaluation of patients with metastatic cancer of known type. In both groups, the goal of the initial evaluation is to determine optimal site-specific first-line treatment after fully characterizing the tumor type and evaluating for specific molecular subsets. For many patients with CUP, site-specific therapy differs markedly from empiric CUP chemotherapy; differences include selection of first-line chemotherapy regimens, use of targeted therapy for identified actionable mutations, and therapy of proven efficacy beyond first-line use.
Fig. 1: Management of Disease in Patients with Metastatic Cancer
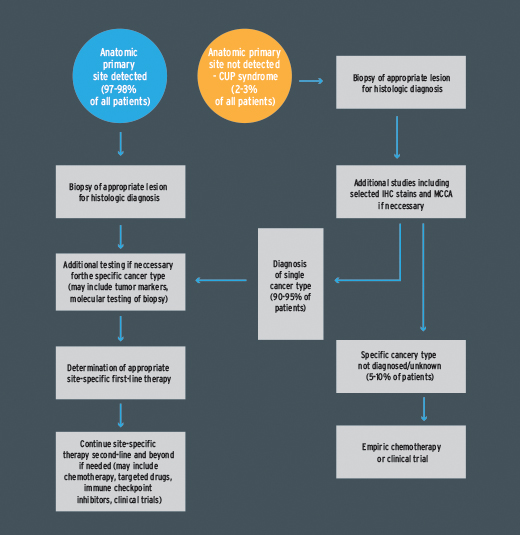
Abbreviations: CUP, cancer of unknown primary site; IHC, immunohistochemistry; MCCA, molecular cancer classifier assay.
The role of comprehensive molecular profiling in the management of CUP is certain to increase in the future. Although therapeutic data are still limited in CUP, identification of a molecular abnormality known to be targetable across multiple tumor types (e.g., HER2, BRAF, EGFR, high MSI, high tumor mutation burden) should lead to strong consideration of treatment with an appropriate targeted therapy, either as first-line (when other options appear unlikely to be beneficial) or subsequent treatment. However, recent suggestions that optimum therapy for CUP can be administered using only the results of comprehensive molecular profiling (i.e., knowledge of the primary site is irrelevant) are premature. At present, few targeted drugs are recommended for first-line single-agent treatment in any solid tumor type. Combination chemotherapy still plays an important role in the treatment of many cancers; few oncologists would recommend the same chemotherapy for patients with breast compared wth colon cancer, nor would they treat the large majority of patients with either of these cancer types with first-line single-agent targeted therapy. Likewise, treatment of a patient with BRAF V600E–mutated cancer with current BRAF-targeted agents would be inappropriate if the primary site was known to be colorectal.
In the past, the heterogeneity of patients with CUP (including different clinical features and diverse cancer types) was an impediment to performing clinical trials and developing effective treatment. The approaches to cancer therapy are now focused on molecular cancer mechanisms, so the heterogeneity of CUPs likely will provide greater opportunities to identify treatable subsets. Additional experience with comprehensive molecular profiling, targeted treatment, and immunotherapy is essential in this patient population.
Dr. Hainsworth is a co-founder and principal investigator at Sarah Cannon Research Institute and a medical oncologist at Tennessee Oncology, PLLC. Dr. Greco is a co-founder at Sarah Cannon Research
Institute and a medical oncologist at Tennessee Oncology, PLLC.
References
- Nystrom JS, Weiner JM, Heffelfinger-Juttner J, et al. Semin Oncol. 1977;4:53-58.
- Pentheroudakis G, Golfinopoulos V, Pavlidis N. Eur J Cancer. 2007;43:2026-2036.
- Greco FA, Hainsworth JD. Cancer of Unknown Primary Site. In DeVita VT Jr, Lawrence TS, Rosenberg SA (eds). Principles and Practice of Oncology (ed 10). Philadelphia, PA: Wolters Kluwer, 2015; 1720-1737.
- Greco FA, Pavlidis N. Semin Oncol. 2009;36:65-74.
- Huebner G, Link H, Kohne CH, et al.; German CUP Study Group. Br J Cancer. 2009;100:44-49.
- Palmeri S, Lorusso V, Palmeri L, et al.; Carcinomas of Unknown Primary Italian Study Group. Cancer. 2006;107:2898-2905.
- Oien KA. Semin Oncol. 2009;36:8-37.
- Greco FA, Lennington WJ, Spigel DR, et al. J Natl Cancer Inst. 2013;105:782-790.
- Weiss LM, Chu P, Schroeder BE, et al. J Mol Diagn. 2013;15:263-269.
- Morawietz L, Floore A, Stork-Sloots L, et al. Virchows Arch. 2010;456:23-29.
- Su AI, Welsh JB, Sapinoso LM, et al. Cancer Res. 2001;61:7388-7393.
- Golub TR, Slonim DK, Tamayo P, et al. Science. 1999;286:531-537.
- Erlander MG, Ma XJ, Kesty NC, et al. J Mol Diagn. 2011;13:493-503.
- Greco FA, Spigel DR, Yardley DA, et al. Oncologist. 2010;15:500-506.
- Meiri E, Mueller WC, Rosenwald S, et al. Oncologist. 2012;17:801-812.
- Handorf CR, Kulkarni A, Grenert JP, et al. Am J Surg Pathol. 2013;37:1067-1075.
- Greco FA, Lennington WJ, Spigel DR, et al. Mol Diagn Ther. 2015;19:91-97.
- Hainsworth JD, Rubin MS, Spigel DR, et al. J Clin Oncol. 2013;31:217-223.
- Moran S, Martínez-Cardús A, Sayols S, et al. Lancet Oncol. 2016;17:1386-1395.
- Yoon HH, Foster NR, Meyers JP, et al. Ann Oncol. 2016;27:339-344.
- Hainsworth JD, Schnabel CA, Erlander MG, et al. Clin Colorectal Cancer. 2012;11:112-118.
- Greco FA, Lennington WJ, Spigel DR, et al. J Cancer Ther. 2012;3:37-43.
- Varadhachary GR, Raber MN, Matamoros A, et al. Lancet Oncol. 2008;9:596-599.
- Hainsworth JD, Spigel DR, Greco FA, et al. J Clin Oncol. 2013;31 (suppl; abstr e15501)
- Hainsworth JD, Greco FA. Drugs Real World Outcomes. 2016;3:115-120.
- Yamada T, Ohtsubo K, Nanjo S, et al. Gan To Kaguka Ryoho. 2012;39:1291-1294.
- Palma NA, Ali SM, O’Connor J, et al. Case Rep Oncol. 2014;7:503-508.
- Tan DS, Montoya J, Ng QS, et al. J Clin Oncol. 2013;31:e237-e239.
- Boku S, Takase N, Onoe T, et al. Austin J Pulm Respir Med. 2015;2:id1030.
- Chung JH, Ali SM, Davis J, et al. Case Rep Oncol. 2014;7:628-632.
- Kato S, Krishnamurthy N, Banks KC, et al. Cancer Res. 2017;77:4238-4246.
- Groschel S, Bommer M, Hutter B, et al. Mol Case Studies. 2016;2:1a001180.
- Hainsworth JD, Meric-Bernstam F, Swanton C, et al. J Clin Oncol. 2018;36:536-542.
- Ross JS, Wang K, Gay L, et al. JAMA Oncol. 2015;1:40-49.
- Hyman DM, Puzanov I, Subbiah V, et al. N Engl J Med. 2015;373:726-736.
- Ross JS, Ali SM, Fasan O, et al. Oncologist. 2017;22:1444-1450.
- Chapman PB, Hauschild A, Robert C, et al.; BRIM-3 Study Group. N Engl J Med. 2011;364:2507-2516.
- Kopetz S, Desai J, Chan E, et al. J Clin Oncol. 2015;33:4032-4038.
- Le DT, Uram JN, Wang H, et al. N Engl J Med. 2015;372:2509-2520.
- Hyman DM, Laetsch TW, Kummar S, et al. J Clin Oncol. 2017;35 (suppl; abstr LBA2501).
- Gatalica Z, Millis SZ, Vranic S, et al. Oncotarget. 2014;5:12440-12447.
- Goodman AM, Kato S, Bazhenova L, et al. Mol Cancer Ther. 2017;16:2598-2608.
- Gay LM, Fabrizio D, Frampton GM, et al. J Clin Oncol. 2017;35 (suppl; abstr 3039).